cath-resolve-hits
A fast, effective way to collapse a list of domain matches to your query sequence(s) down to the best, non-overlapping subset (ie domain architecture).
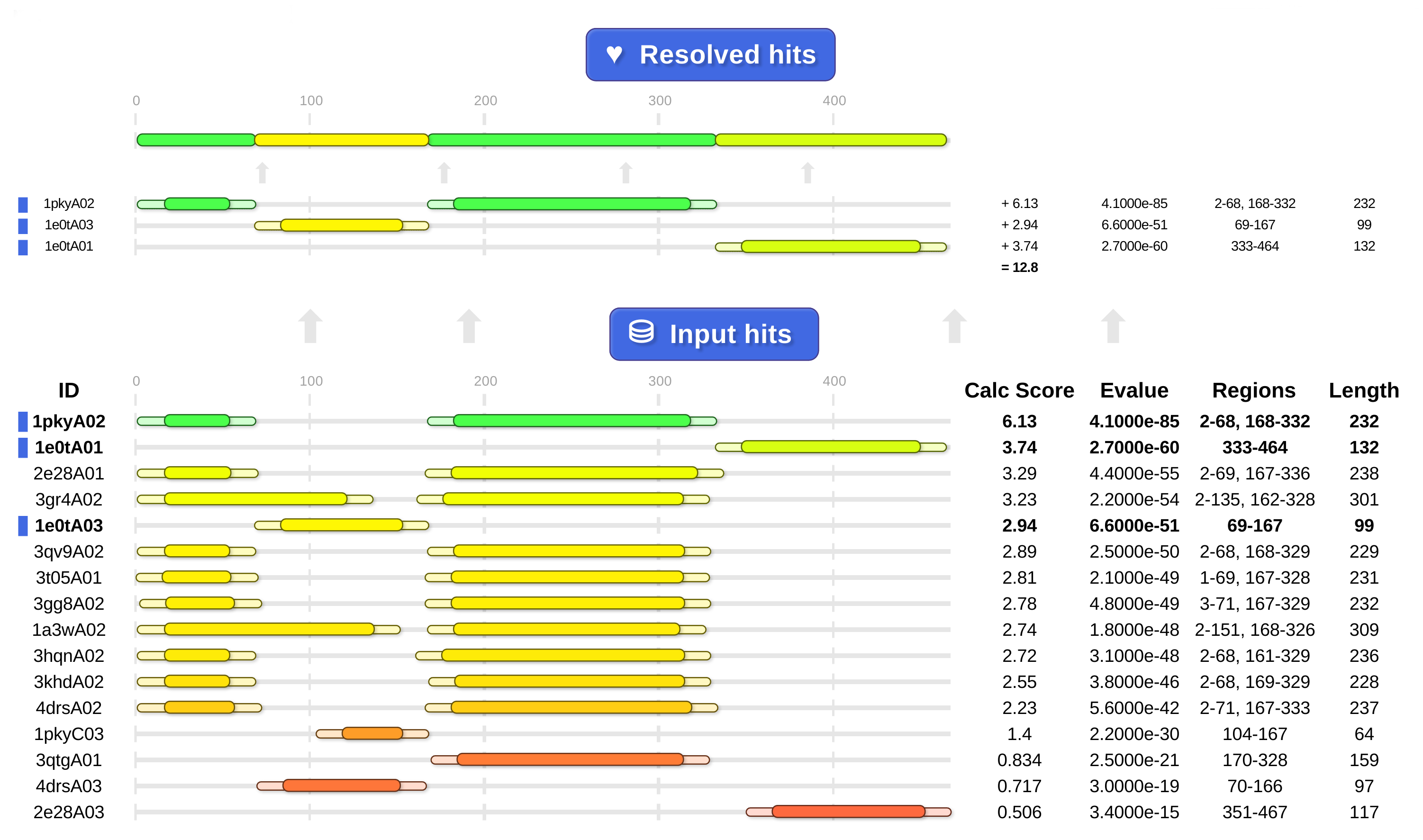
Above: cath-resolve-hits' HTML output shows it choosing the hits at the top from the list of candidates at the bottom
Features
Fast
- Can process around 1–2 million input hits per second
Powerful
- Finds the optimal result that maximises the sum of hits' scores
- Handles discontinuous domains
- Supports tolerance for overlaps between hits; auto-resolves any that occur
Transparent
- Provides visualisation of input data and decisions via graphical HTML
Simple
- Outputs in plain text or JSON
- Uses a simple default input file format
- Also accepts HMMER domtblout files and hmmsearch output files
- Accepts input that hasn't been pre-sorted or even pre-grouped (but can exploit pre-grouping where specified)
Configurable
- Allows users to determine their own scoring system to be maximised
- Offers many easy-to-use options to configure the default behaviour
Getting started
At present, cath-resolve-hits only requires one argument: a file which, by default, contains lines like:
qyikaz 1mkfA01/12-210-i5_1,2.9e-20 2983.29780221221 3-103
qyikaz 1mkfA01/12-210-i5_2,4.9e-19 3510.41568607646 101-224
qyikaz 1mkfA01/12-210-i5_3,7e-25 3552.10980383852 825-928
qyikaz 1mkfA01/12-210-i5_4,3.5e-15 2470.04912752062 953-1053
Where the fields are space-separated and are:
- query_protein_id : An identifier for the query protein sequence
- match_id : An identifier for the match
- score : A (strictly positive) score indicating how good it would be to have that hit in the final result
- starts_stops : The starts/stops on the query sequence, given in the format: 37–124,239–331
The output is the non-overlapping subset that maximises the sum of the hits' scores.
The input file can contain data for multiple different query protein sequences and they needn't be grouped into consecutive lines (though if they are, specify the --input-hits-are-grouped option).
Usage
The current full --help usage information is:
Usage: cath-resolve-hits [options] <input_file>
Collapse a list of domain matches to your query sequence(s) down to the
non-overlapping subset (ie domain architecture) that maximises the sum of the
hits' scores.
When <input_file> is -, the input is read from standard input.
The input data may contain unsorted hits for different query protein sequences.
However, if your input data is already grouped by query protein sequence, then
specify the --input-hits-are-grouped flag for faster runs that use less memory.
Miscellaneous:
-h [ --help ] Output help message
-v [ --version ] Output version information
Input:
--input-format <format> (=raw_with_scores) Parse the input data from <format>, one of available formats:
hmmer_domtblout - HMMER domtblout format (must assume all hits are continuous)
hmmscan_out - HMMER hmmscan output format (can be used to deduce discontinuous hits)
hmmsearch_out - HMMER hmmsearch output format (can be used to deduce discontinuous hits)
raw_with_scores - "raw" format with scores
raw_with_evalues - "raw" format with evalues
--min-gap-length <length> (=30) When parsing starts/stops from alignment data, ignore gaps of less than <length> residues
--input-hits-are-grouped Rely on the input hits being grouped by query protein
(so the run is faster and uses less memory)
Segment overlap/removal:
--overlap-trim-spec <trim> (=30/10) Allow different hits' segments to overlap a bit by trimming all segments using spec <trim>
of the form n/m (n is a segment length; m is the *total* length to be trimmed off both ends)
For longer segments, total trim stays at m; for shorter, it decreases linearly (to 0 for length 1).
To choose: set m to the biggest total trim you'd want for a really long segment;
then, set n to length of the shortest segment you'd want to have that total trim
--min-seg-length <length> (=7) Ignore all segments that are fewer than <length> residues long
Hit preference:
--long-domains-preference <val> (=0) Prefer longer hits to degree <val>
(<val> may be negative to prefer shorter; 0 leaves scores unaffected)
--high-scores-preference <val> (=0) Prefer higher scores to degree <val>
(<val> may be negative to reduce preference for higher scores; 0 leaves scores unaffected)
--apply-cath-rules [DEPRECATED] Apply rules specific to CATH-Gene3D during the parsing and processing
--naive-greedy Use a naive, greedy approach to resolving (not recommended except for comparison)
Hit filtering:
--worst-permissible-evalue <evalue> (=0.001) Ignore any hits with an evalue worse than <evalue>
--worst-permissible-bitscore <bitscore> (=10) Ignore any hits with a bitscore worse than <bitscore>
--worst-permissible-score <score> Ignore any hits with a score worse than <score>
--filter-query-id <id> Ignore all input data except that for query protein(s) <id>
(may be specified multiple times for multiple query proteins)
--limit-queries [=<num>(=1)] Only process the first <num> query protein(s) encountered in the input data
Output ([...]-to-file options may be specified multiple times):
--hits-text-to-file <file> Write the resolved hits in plain text to file <file>
--quiet Suppress the default output of resolved hits in plain text to stdout
--output-trimmed-hits When writing out the final hits, output the hits' starts/stop as they are *after trimming*
--summarise-to-file <file> Write a brief text summary of the input data to file <file> (or '-' for stdout)
--html-output-to-file <file> Write the results as HTML to file <file> (or '-' for stdout)
--json-output-to-file <file> Write the results as JSON to file <file> (or '-' for stdout)
--export-css-file <file> Export the CSS used in the HTML output to <file> (or '-' for stdout)
HTML:
--restrict-html-within-body Restrict HTML output to the contents of the body tag.
The contents should be included inside a body tag of class crh-body
--html-max-num-non-soln-hits <num> (=80) Only display up to <num> non-solution hits in the HTML
--html-exclude-rejected-hits Exclude hits rejected by the score filters from the HTML
Detailed help:
--cath-rules-help Show help on the rules activated by the (DEPRECATED) --apply-cath-rules option
--raw-format-help Show help about the raw input formats (raw_with_scores and raw_with_evalues)
The standard output is one line per selected hit, preceded by header lines (beginning "#"), the last of which (beginning "#FIELDS") lists the fields in the file, typically:
#FIELDS query-id match-id score boundaries resolved
(`boundaries` and `resolved` describe a domain's starts / stops; `resolved` may include adjustments made to resolve overlaps between hits)
Please tell us your cath-tools bugs/suggestions : https://github.com/UCLOrengoGroup/cath-tools/issues/new
Output format
The standard output is one line per selected hit, preceded by header lines (beginning #), the last of which (beginning #FIELDS) lists the fields in the file, typically:
#FIELDS query-id match-id score boundaries resolved
(boundaries and resolved describe a domain's starts / stops; resolved may include adjustments made to resolve overlaps between hits)
Alternatively, consider --json-output or --html-output.
Warning
For now, don't set --high-scores-preference too high (say, above 3.5) because otherwise the range of scores will exceed what can be reliably handled by a 32-bit floating point number.
How Fast?
To give a very rough idea: on an SSD-enable laptop, we've seen cath-resolve-hits process some large data files at around 1–2 million hits per second. That test setup was probably a bit unrealistic so your mileage may vary significantly. For reference: the GCC build appeared to run quite a bit faster than the Clang build.
CATH Rules invoked by option --apply-cath-rules
The --apply-cath-rules option applies the following CATH–Gene3D specific rules when parsing from hmmer_domtmblout or hmmsearch_out format files.
Discontinuous domains
If hit's match ID is like dc_72a964d791dea7a3dd35a8bbf49385b8 (matches /^dc_\w{32}$/), then:
- use the
ali_from/ali_tofields rather thanenv_from/env_toto determine the final start/stop and - ignore gaps when parsing an alignment from a hmmsearch_outfile (ie keep the hit as one continuous segment).
Bitscore reductions
If the conditional-evalue is ≤ 0.001 but the independent-value is > 0.001, then quarter the bitscore when parsing the hit.
Feedback
Please tell us about your cath-tools bugs/suggestions here.