cath-superpose
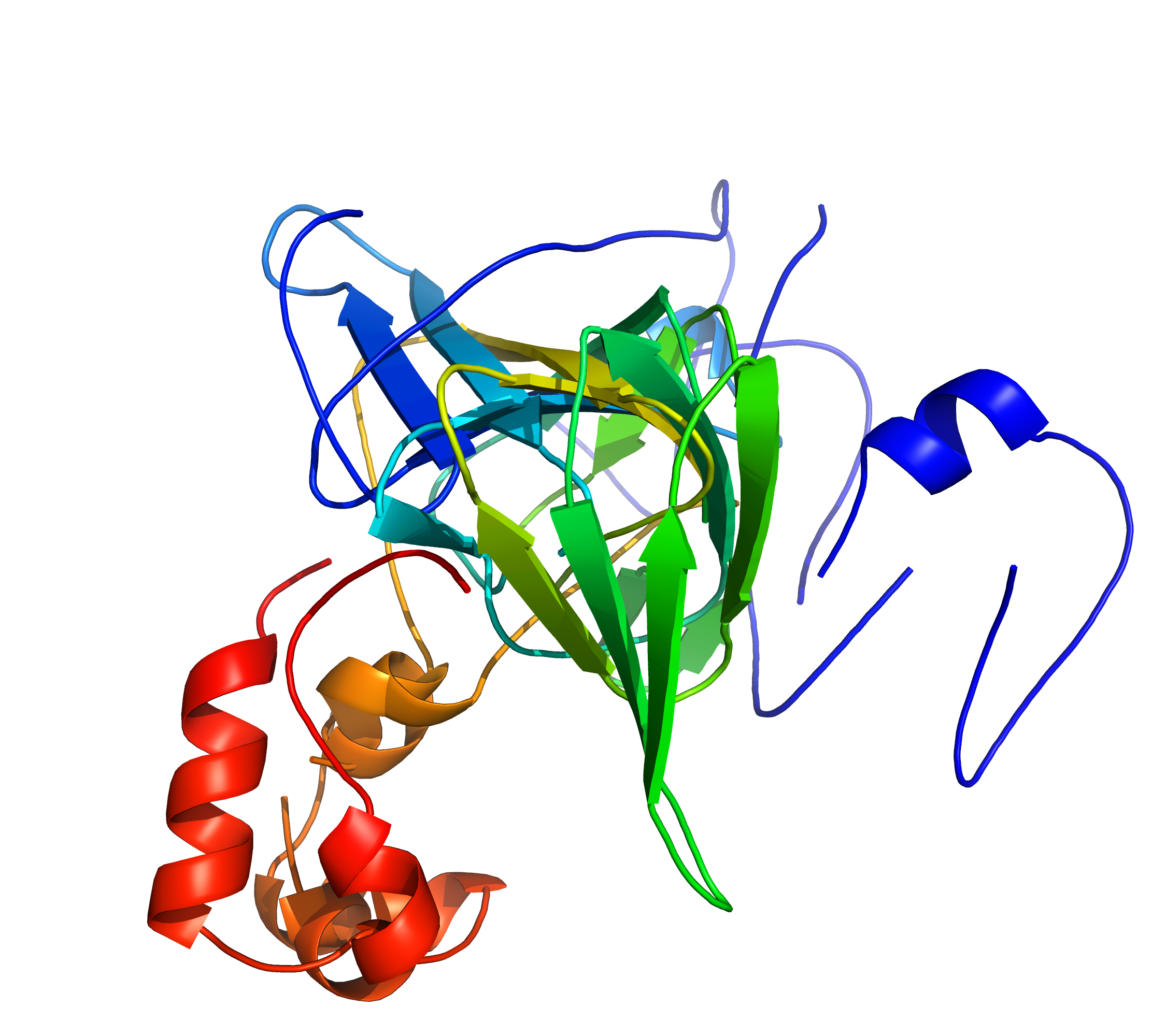
Left: A standard superposition of 1fi2A00/1j58A01 is disrupted by divergent regions;
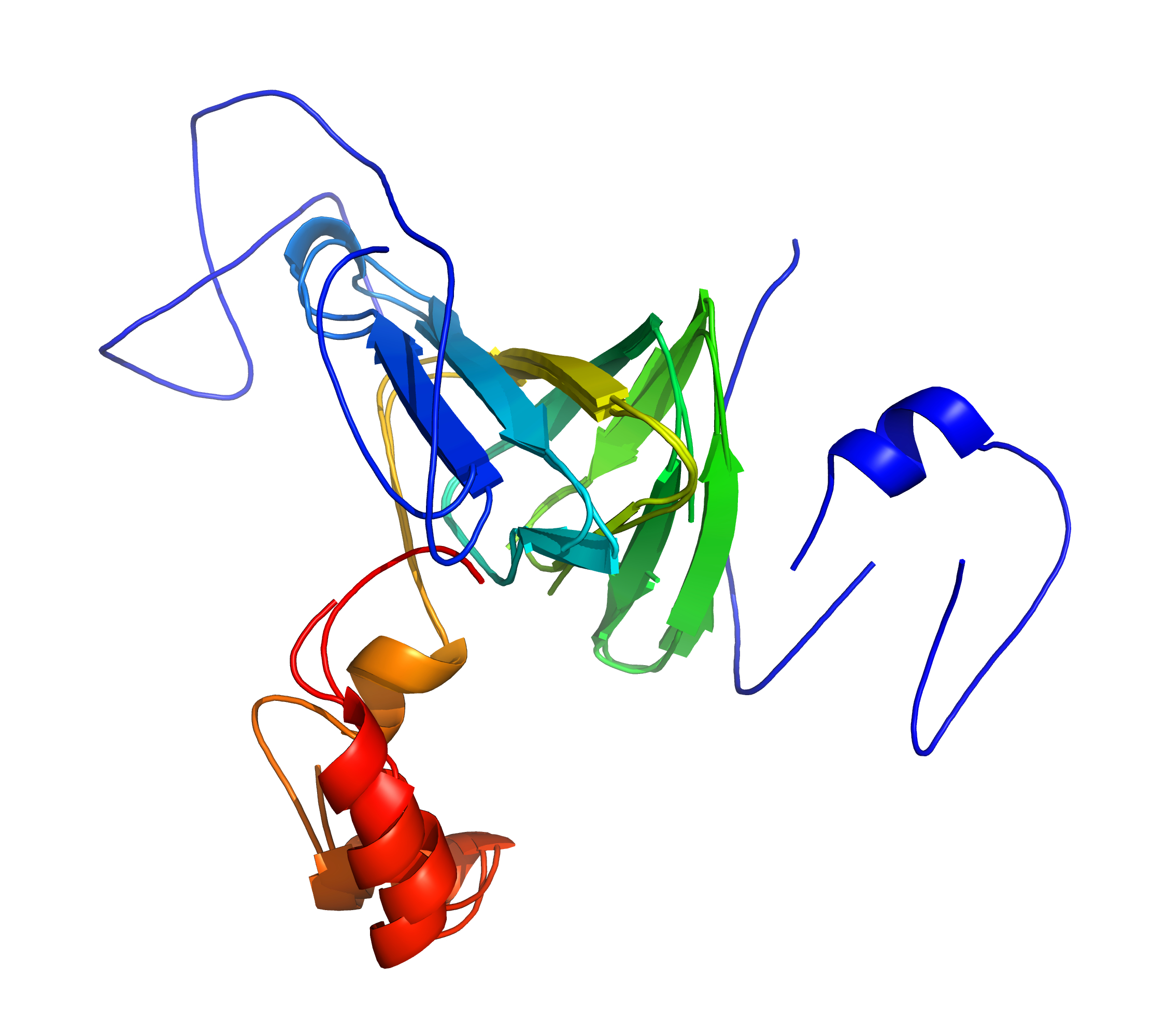
Right: cath-superpose does better by targeting the similar regions (but at the cost of increased RMSD). [rendered by PyMOL]
Introduction
The cath-superpose tool makes superpositions that look better (even though they may have higher overall RMSDs).
It does this by focusing the superposition on those parts of the alignment that align well so that other aligned regions with greater variance don't disrupt the superposition.
Usage Overview
cath-superpose needs an alignment of the structures to use to perform the superposition. You can provide an alignment that cath-superpose should use, which at present can be any of:
- a SSAP alignment (ie
.listfile) (seecath-ssap) - a FASTA alignment
- a CORA alignment
- a file containing all the pairwise SSAP scores between a group of structures in a directory contains all the corresponding SSAP alignment files
- the rule to just align residues by matching their names (number+insert) (useful for superposing models of the same protein)
...or by default, it'll use the --do-the-ssaps option, which means it gets its alignment by performing the all-vs-all pairwise cath-ssaps in a temporary directory (or one you specify) and then gluing those cath-ssap alignments together.
Example: to superpose two structures, you might use commands like:
export CATH_TOOLS_PDB_PATH=/global/data/directories/pdb
cath-superpose --pdb-infile $CATH_TOOLS_PDB_PATH/1c0pA01 --pdb-infile $CATH_TOOLS_PDB_PATH/1hdoA00
(With no output specified, this will use the default of firing up PyMOL to view the superposition; see the usage for different options.)
Alignment refining
When cath-superpose is gluing pairwise alignments together (under --ssap-scores-infile or --do-the-ssaps), it may refine the alignments according to the --align-refining option. However cath-superpose won't change a complete alignment that you give it under the other options. If you want to refine your alignment, please try cath-refine-align instead.
Multiple Superpositions
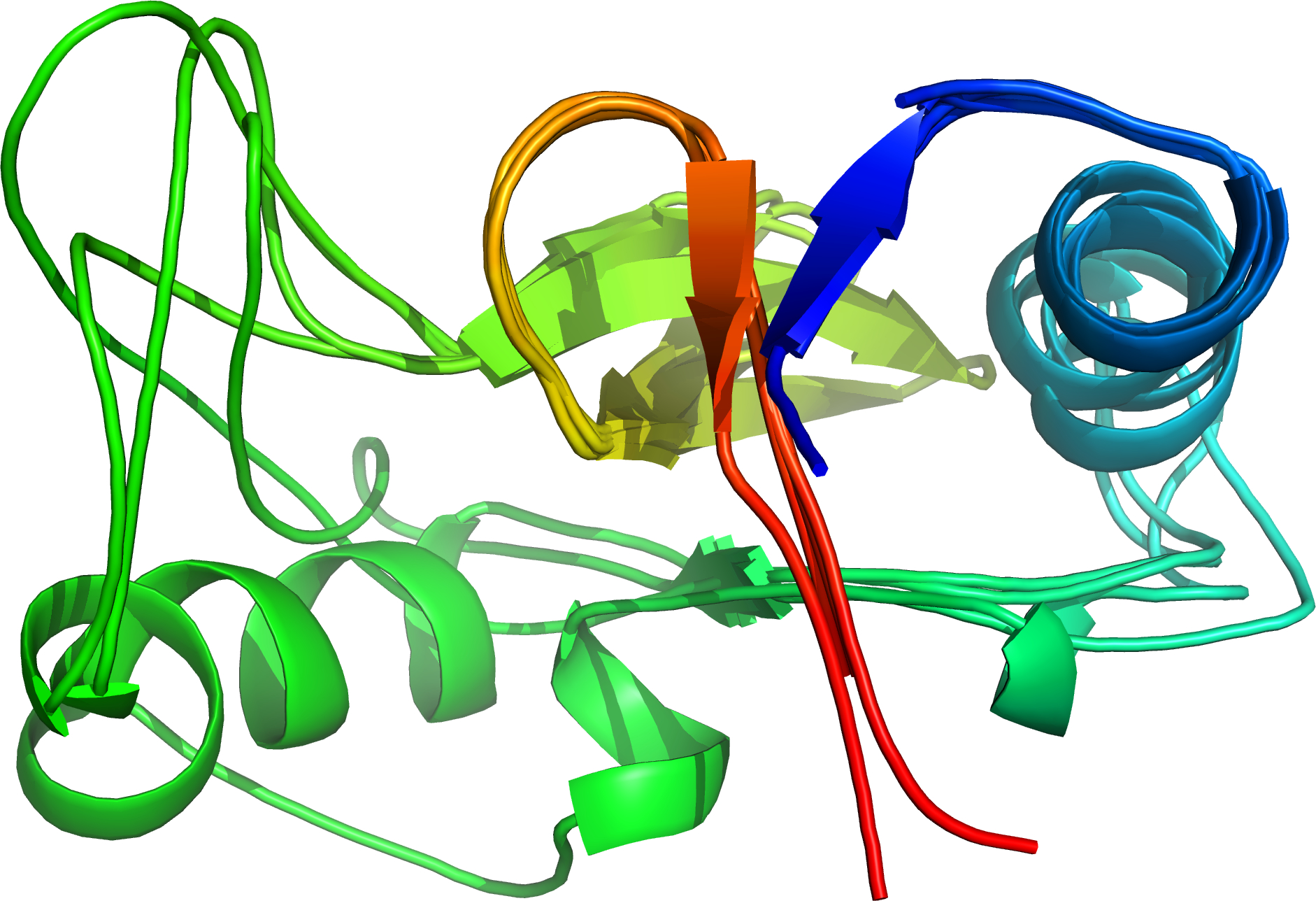
Four similar structures superposed by cath-superpose [rendered by PyMOL]
cath-superpose can superpose more than two structures by combining results from pairwise cath-ssaps. This previously required you to use a separate script to prepare the data but cath-superpose will now default to the --do-the-ssaps option, which performs the necessary cath-ssaps for you. Just make sure you configure your environment variables so that cath-ssap can find the input files.
You can also specify corresponding pairs of structures and the regions of the structures that you want to align and superpose:
cath-superpose --pdb-infile 2vxn --align-regions 'D[2vxnA00]2-250:A' --pdb-infile 2y7e --align-regions 'D[2y7eB00]-2-275:B' --pdb-infile 3b4u --align-regions 'D[3b4uA00]4-290:A' --pdb-infile 1p1x --align-regions 'D[1p1xB00]1000-1250:B'
The format is like: D[5inwB02]251-348:B,408-416A:B. Put
Usage
The current usage information is:
Usage: cath-superpose alignment_source pdb_file_source [superposition_outputs]
Superpose protein structures using an existing alignment
Please specify:
* at most one superposition JSON or alignment (default: --do-the-ssaps)
* one method of reading PDB files (number to match the alignment)
PyMOL is started if no alignment or superposition output option is specified
Miscellaneous:
-h [ --help ] Output help message
-v [ --version ] Output version information
Input:
Alignment source:
--res-name-align Align residues by simply matching their names (numbers+insert)
(for multiple models of the same structure)
--fasta-aln-infile <file> Read FASTA alignment from file <file>
--ssap-aln-infile <file> Read SSAP alignment from file <file>
--cora-aln-infile <file> Read CORA alignment from file <file>
--ssap-scores-infile <file> Glue pairwise alignments together using SSAP scores in file <file>
Assumes all .list alignment files in same directory
--do-the-ssaps [=<dir>(="")] Do the required SSAPs in directory <dir>; use results as with --ssap-scores-infile
Use a suitable temp directory if none is specified
Alignment refining:
--align-refining <refn> (=NO) Apply <refn> refining to the alignment, one of available values:
NO - Don't refine the alignment
LIGHT - Refine any alignments with few entries; glue alignments one more entry at a time
HEAVY - Perform heavy (slow) refining on the alignment, including when gluing alignments together
This can change the method of gluing alignments under --ssap-scores-infile and --do-the-ssaps
Superposition source:
--json-sup-infile <file> Read superposition from file <file>
ID options:
--id arg Structure ids
PDB files source:
--pdb-infile <pdbfile> Read PDB from file <pdbfile> (may be specified multiple times)
--pdbs-from-stdin Read PDBs from stdin (separated by line: "END ")
Regions:
--align-regions <regions> Handle region(s) <regions> as the alignment part of the structure.
May be specified multiple times, in correspondence with the structures.
Format is: D[5inwB02]251-348:B,408-416A:B
(Put <regions> in quotes to prevent the square brackets confusing your shell ("No match"))
Output:
Alignment output:
--aln-to-cath-aln-file arg [EXPERIMENTAL] Write the alignment to a CATH alignment file
--aln-to-cath-aln-stdout [EXPERIMENTAL] Print the alignment to stdout in CATH alignment format
--aln-to-fasta-file arg Write the alignment to a FASTA file
--aln-to-fasta-stdout Print the alignment to stdout in FASTA format
--aln-to-ssap-file arg Write the alignment to a SSAP file
--aln-to-ssap-stdout Print the alignment to stdout as SSAP
--aln-to-html-file arg Write the alignment to a HTML file
--aln-to-html-stdout Print the alignment to stdout as HTML
Superposition output:
--sup-to-pdb-file arg Write the superposed structures to a single PDB file arg, separated using faked chain codes
--sup-to-pdb-files-dir arg Write the superposed structures to separate PDB files in directory arg
--sup-to-stdout Print the superposed structures to stdout, separated using faked chain codes
--sup-to-pymol Start up PyMOL for viewing the superposition
--pymol-program arg (="pymol") Use arg as the PyMOL executable for viewing; may optionally include the full path
--sup-to-pymol-file arg Write the superposition to a PyMOL script arg
(Recommended filename extension: .pml)
--sup-to-json-file arg Write the superposition to JSON superposition file
(Recommended filename extension: .sup_json)
Viewer (eg PyMOL, Jmol etc) options:
--viewer-colours <colrs> Use <colrs> to colour successive entries in the viewer
(format: colon-separated list of comma-separated triples of RGB values between 0 and 1)
(will wrap-around when it runs out of colours)
--gradient-colour-alignment Colour the length of the alignment with a rainbow gradient (blue -> red)
--show-scores-if-present Show the alignment scores
(use with gradient-colour-alignment)
--scores-to-equivs Show the alignment scores to equivalent positions, which increases relative scores where few entries are aligned
(use with --gradient-colour-alignment and --show-scores-if-present)
--normalise-scores When showing scores, normalise them to the highest score in the alignment
(use with --gradient-colour-alignment and --show-scores-if-present)
Superposition content:
--regions-context <context> (=alone) Show the alignment regions in the context <context>, one of available options:
alone - alone
in_chain - within the chain(s) in which the regions appear
in_pdb - within the PDB in which the regions appear
--show-dna-within-dist <dist> (=4) Show DNA within <dist>Å of the alignment regions
--show-organic-within-dist <dist> (=10) Show organic molecules within <dist>Å of the alignment regions
Usage examples:
* cath-superpose --ssap-aln-infile 1cukA1bvsA.list --pdb-infile $PDBDIR/1cukA --pdb-infile $PDBDIR/1bvsA --sup-to-pymol
(Superpose 1cukA and 1bvsA (in directory $PDBDIR) based on SSAP alignment file 1cukA1bvsA.list and then display in PyMOL)
* cat pdb1 end_file pdb2 end_file pdb3 | cath-superpose --pdbs-from-stdin --sup-to-stdout --res-name-align
(Superpose the structures from stdin based on matching residue names and then write them to stdout [common Genome3D use case])
Please tell us your cath-tools bugs/suggestions : https://github.com/UCLOrengoGroup/cath-tools/issues/new
Feedback
Please tell us about your cath-tools bugs/suggestions here.